• Mona Karazivan, DMD, M.Sc. •
• Kevork Manoukian, DMD, MBA •
• BenoÓt Lalonde, DMD, MSD, FRCD(C) •
Gardner syndrome is a type of hereditary gastrointestinal polyposis. Dental professionals should be aware that this syndrome can present as multiple impacted teeth and sometimes as large osteomas in the head and neck area.
Following a brief review of literature, we present two cases of Gardner syndrome. One of these cases was diagnosed after a dental examination. The high incidence of malignant transformation of polyps into colorectal cancer indicates the importance of early diagnosis and follow-up.
MeSH Key Words: Gardner syndrome; jaw neoplasm; tooth abnormalities
© J Can Dent Assoc 2000; 66:26-30
Le syndrome de Gardner ou polypose adťnomateuse familiale (PAF) reprťsente une forme de polypose co lique. Entre 1950 et 1954, Gardner et ses collaborateurs1,2 dťcrivaient un syndrome associant des ostťomes multiples (surtout des os faciaux), des kystes ťpidermoÔdes, des fibromes de la peau, des polypes intestinaux et des tumeurs desmoÔdes. Plus tard, on associa ťgalement ŗ ce syndrome la possibilitť de lipomes, de lťiomyomes et d’odontomes3.
…tiologie
Le syndrome de Gardner est causť principalement par une mutation du gŤne
Adenomatous polyposis coli (APC). Ce gŤne, identifiť au dťbut des annťes 90 par
plusieurs ťquipes de chercheurs (Groden et coll., Joslyn et coll., Kinzler et coll.,
Nishisho et coll.), se trouve sur le bras long du chromosome numťro 54-7.
Jusqu’ŗ prťsent, plus de 200 mutations ont ťtť rapportťes. La position de la
mutation au niveau de la sťquence du code du gŤne APC peut influencer l’expression
du phťnotype8.
Ces mutations ou anormalitťs gťnťtiques sont habituel lement transmises par un des parents, mais peuvent survenir de novo dans un tiers des cas. La transmission est autosomique dominante, la pťnťtrance, trŤs ťlťvťe (de 80 ŗ 100 %) et l’expressivitť, trŤs variable. Le syndrome de Gardner est trŤs rare, touchant 1 sur 14,000 personnes9.
Manifestations cliniques
Au cours des annťes 1950, Gardner nota que plusieurs patients souffrant de
polypose familiale du colon ťtaient aussi atteints de lťsions extracoliques (p. ex.,
ostťomes multiples et tumeurs des tissus mous). Or, on dťfinit maintenant mieux ses
signes et ses symptŰmes cliniques.
Neurologie
Au niveau du systŤme nerveux central, on remarque chez certains patients
des degrťs variables de retard mental.
FaciŤs
Des ostťomes de dimensions variables peuvent apparaÓtre au visage, soit
ŗ la mandibule, au maxillaire ou au niveau des os frontaux et des sinus ťthmoÔdes.
Certains patients peuvent souffrir d’une fissure palatine et labiale ou d’une
luette bifide.
Dermatologie
Au niveau cutanť, environ la moitiť des patients souffrant du syndrome de
Gardner prťsentent des kystes ťpidermoÔdes, qui peuvent apparaÓtre n’importe oý
sur la tÍte, le visage, le tronc et mÍme les jambes. La prťsence de tels kystes devrait
susciter un examen de dťpistage du syndrome9. Ces kystes apparaissent
gťnťralement vers l’‚ge de 13 ans, soit avant l’apparition des polypes
intestinaux. Enfin, de nouveaux kystes peuvent apparaÓtre pťriodiquement.
En plus des polypes intestinaux, on observe l’apparition de tumeurs fibreuses de la peau, comme les fibromes et les tumeurs desmoÔdes. La tumeur desmoÔde, retrouvťe chez en viron 10 % des patients souffrant du syndrome de Gardner, survient en grande partie au niveau de la cicatrice abdominale lorsqu’il y a eu rťsection du colon. Elle peut par contre survenir en l’absence de chirurgie. Des lipomes ou lipofibromes de la peau peuvent aussi Ítre observťs chez ces patients7,10.
SystŤme gastro-intestinal et structures environnantes
La maladie est caractťrisťe par la prťsence de polypes adťnomateux au
niveau du colon et du rectum. On peut en noter aussi peu qu’une centaine et autant
que quelques milliers7. Ces polypes ont dťfinitivement le potentiel de
progresser en carcinome du colon, ce qui nous porte ŗ croire que le gŤne APC joue un
rŰle inhibiteur important dans le dťveloppement du cancer du colon. La portion terminale
dťficiente du chromosome 5, oý se retrouve le gŤne APC, expliquerait la propension ŗ
dťvelopper un cancer chez les patients souffrant du syndrome de Gardner. Cette propension
pourrait trŤs bien s’expliquer par le fait que ces patients ont un nombre trŤs
ťlevť de polypes adťnomateux et seraient donc jusqu’ŗ quelques milliers de fois
plus ŗ risque qu’un individu n’en ayant qu’un11.
Les polypes extracoloniques sont frťquemment observťs sous forme de polypes du systŤme gastro-intestinal supťrieur (exception faite de l’oesophage)12. Ces polypes sont de deux types histologiquement distincts : les polypes glandulaires fundiques et les polypes adťnomateux. Bien que plus communs, les polypes glandulaires fundiques sont en fait des hamartomes et n’ont aucun potentiel malin. Par contre, les polypes adťnomateux gastriques, plus rares, sont potentiel lement malins et tendent ŗ toucher davantage les Japonais7.
Les polypes apparaissent habituellement entre 13 et 25 ans et sont associťs ŗ un risque extrÍmement ťlevť (presque 100 %) de transformation maligne s’ils ne sont pas traitťs13.
Une autre particularitť du syndrome de Gardner est la prťsence de tumeurs desmoÔdes, qualifiťes de fibromatoses agressives. Celles-ci dťcoulent principalement des cellules mťsenchymateuses comme celles situťes au niveau des aponťvroses musculaires et des fascias. Malgrť une infiltration destructrice des tissus environnants, les tumeurs desmoÔdes sont gťnťralement considťrťes comme des tumeurs fibreuses bťnignes qui ne produisent pas de mťtastases. Elles sont composťes de cellules fibroblastiques matures et n’ont pas de capsule vťritable. La pathogenŤse de ces tumeurs demeure incertaine. Par contre, des facteurs traumatiques, gťnťtiques et endocriniens ont ťtť identifiťs comme pouvant jouer un rŰle14.
Les tumeurs desmoÔdes peuvent apparaÓtre ŗ diffťrents endroits du corps, mais se trouvent principalement dans la rťgion abdominale, ŗ savoir le mťsentŤre du petit intestin ou la cavitť abdominale. Malheureusement, malgrť des caractťristiques histologiques bťnignes, les rťcidives locales sont frťquentes (65 %) aprŤs une excision chirurgicale15. Si on ne les traite pas, ces tumeurs, trois fois plus communes chez les femmes que chez les hommes, peuvent mener ŗ une morbiditť locale voire ŗ la mort.
Enfin, on a observť des lťiomyomes dans la rťgion rťtropťritonťale et au niveau de l’estomac chez les patients atteints du syndrome de Gardner.
SystŤme endocrinien
L’incidence prťcise des nťoplasies endocriniennes chez les patients
souffrant du syndrome de Gardner est inconnue, car celles-ci peuvent Ítre non
fonctionnelles ou occultes. Le carcinome papillaire de la thyroÔde est la nťoplasie la
plus commune chez ces patients. Le ratio femme/homme dans ces cas est de 17 pour 1. La
dťtection se fait habituellement vers l’‚ge de 30 ans. Enfin, ces tumeurs sont
souvent multifocales16.
SystŤme bucco-dentaire
Comme nous l’avons dťjŗ mentionnť, des ostťomes multiples
(exostoses et ťnostoses) peuvent apparaÓtre un peu partout au visage, affectant surtout
l’os frontal, le maxillaire, l’angle de la mandibule et le bord infťrieur de la
mandibule. Un ostťome localisť ŗ l’angle de la mandibule est hautement
caractťristique du syndrome de Gardner7. Au microscope, cet os est mature et
consiste en des systŤmes de Havers bien dťveloppťs. Ce syndrome est aussi associť ŗ
de multiples dents surnumťraires incluses et ŗ des odontomes complexes (surtout prŤs
des racines des prťmolaires). Souvent, l’analyse de ces tissus montre des cellules
au stade actif. Enfin, on observe parfois des kystes dentifŤres (ou folliculaires), de
l’hypercťmentose et des dents surnumťraires. Quant ŗ la protťine APC, on la
trouve ŗ diffťrents stades du cycle de formation de l’odontoblaste17.
Toutes ces anomalies bucco-dentaires sont de 10 ŗ 20 % plus communes chez les patients souffrant du syndrome de Gardner que dans la population en gťnťral, et 50 % de ces patients dťvelopperont au moins l’une de ces anomalies7.
SystŤme oculaire
Une derniŤre caractťristique des patients souffrant du syndrome de
Gardner est la prťsence d’hypertrophie congťnitale de l’ťpithťlium
pigmentaire de la rťtine18. L’examen du fond de l’oeil rťvŤle
qu’environ 90 % de ces patients en souffrent. Ces lťsions maculaires sont
bilatťrales et multiples (de 1 ŗ 30) et constituent un marqueur clinique spťcifique et
sensible du syndrome de Gardner9. Il est ŗ noter ťgalement que certains
patients souffrent d’une atrophie du nerf optique.
CritŤres diagnostics
et traitement
Souvent, ŗ cause de troubles ou de retards psychomoteurs et de
l’apparence faciale particuliŤre, des analyses cytogťnťtiques sont pratiquťes ŗ
un trŤs jeune ‚ge. Par contre, lorsqu’il n’y a pas de retard mental et que les
deux parents sont normaux, la maladie est dťtectťe plus tard, ŗ l’adolescence.
Les radiographies dentaires panoramiques peuvent nous aider ŗ ťtablir un diagnostic si on note des lťsions radiopaques ŗ un ou aux deux maxillaires ou si on observe des dents surnumťraires et/ou incluses. Ces pathologies peuvent Ítre observťes pťriodiquement et ne nťcessitent habituellement pas de chirurgie.
Si on soupÁonne un nodule thyroÔdien ou un autre pro blŤme au niveau de cette glande, il est conseillť d’effectuer des examens de fonction thyroÔdienne, des ultrasons ou une scintigraphie16. L’examen cytologique par biopsie ŗ l’aiguille fine pourra aider ŗ prťciser la prťsence de cellules atypiques.
En gťnťral, lorsqu’on soupÁonne un diagnostique de syndrome de Gardner, une ťvaluation en gastro-entťrologie est indiquťe. La recto-sigmoÔdoscopie aidera ŗ confirmer la prťsence ou l’absence de polypes intestinaux. Ces polypes peuvent causer la diarrhťe, la constipation, des douleurs intestinales ou des saignements rectaux. Si nťcessaire, une biopsie de ces polypes peut Ítre faite. Tel que spťcifiť auparavant, si aucun traitement prophylactique n’est prodiguť, un carcinome du colon et/ou du rectum pourra se dťvelopper chez ces patients vers l’‚ge de 40 ans. Certains auteurs suggŤrent la rťsection du colon dŤs le diagnostic ťtabli, quel que soit l’‚ge du patient7. La colectomie avec anastomose ilťo-rectale semble Ítre le traitement recommandť pour les patients souffrant du syndrome de Gardner avec polypes du colon.
Pour ce qui est des tumeurs desmoÔdes, puisqu’elles sont souvent invasives, l’ablation complŤte peut Ítre trŤs difficile. Certains suggŤrent l’usage de mťdicaments, comme les anti-inflammatoires non stťroÔdiens (sulindac), le tamoxifen ou l’ascorbate, afin de rťduire temporairement le nombre et le volume des polypes15. La chimiothťrapie et la radiothťrapie sont aussi utilisťes. Or, souvent, il est difficile de contrŰler ces tumeurs de nature rťcurrente et d’incidence morbide ou mÍme fatale.
Cas cliniques
Cas no 1
Les deux filles de la patiente ainsi que sa soeur rapportaient les mÍmes problŤmes, et toutes trois ťtaient suivies en gastro-entťrologie. La patiente avait subi une colectomie en 1978. Sa soeur ainsi que ses deux filles, ‚gťes de 30 et 38 ans, avaient subi la mÍme chirurgie.
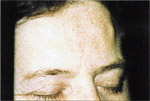
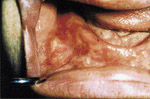
L’examen extra-buccal rťvťla trois exostoses sur le front et une sur la joue droite (ill. 1). L’examen buccal confirma l’ťdentation complŤte et dťcela des lťsions indurťes ŗ diffťrents endroits (ill. 2).
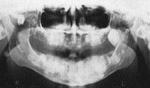
L’examen radiologique (panoramique, cťphalomťtrique ainsi que tomodensitomťtrique) confirma la prťsence d’exostoses spectaculaires atteignant les maxillaires de faÁon gťnťra lisťe. De plus, des ostťomes isolťs furent notťs ŗ l’angle gonial, ŗ l’arcade zygomatique du cŰtť droit et ŗ la tubťrositť gauche (ill. 3). Les sinus ethmoÔdaux et l’os frontal ťtaient ťgalement atteints (ill. 4).
 |
 |
 |
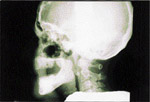 |
| llustration 1 : Prťsence d’ostťomes dans la rťgion frontale. | Illustration 2 : Prťsence d’un ostťome dans la rťgion vestibulaire infťrieure droite causant un dťplacement de la prothŤse complŤte infťrieure. | Illustration 3 : Vue panoramique rťvťlant la prťsence massive d’ostťomes et d’ťnostoses au niveau des maxillaires. On note ťgalement la prťsence d’ostťomes ŗ l’angle gonial, ŗ l’arcade zygomatique du cŰtť droit et ŗ la tubťrositť gauche. | Illustration 4 : Vue cťphalomťtrique. On remarque la prťsence d’ostťomes au niveau frontal et aux sinus ethmoÔdaux. Les lťsions osseuses ŗ l’angle de la mandibule ainsi qu’ŗ l’arcade zygomatique sont ťgalement visibles. |
La patiente fut rťfťrťe en chirurgie maxillo-faciale afin de subir l’ablation des ostťomes qui interfťraient avec la fonction masticatrice. Elle est suivie rťguliŤrement en gastro-entťrologie, et son ťtat est stabilisť.
Cas no 2
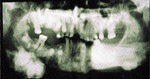
Un patient ‚gť de 33 ans fut rťfťrť ŗ l’un des auteurs (KM) en consultation pour des infections reliťes ŗ des extractions dentaires. Lors de la premiŤre visite, une radiographie panoramique fut prise, qui confirma la prťsence d’exostoses impressionnantes (ill. 5).
 |
 |
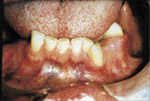 |
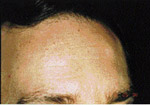
| Illustration 5 : Vue panoramique. On note la prťsence de multiples inclusions dentaires d’odontomes ainsi que des ostťomes gťants au niveau mandibulaire, et ce, bilatťralement. | Illustration 6 : Prťsence d’un ostťome au niveau frontal gauche. | Illustration 7 : La dťformation dans les rťgions sous-maxillaires gauche et droite rťvťla ŗ la palpation des masses indurťes et fixťes au corps de la mandibule. La vue panoramique confirma la prťsence d’ostťomes de dimensions impressionnantes. |
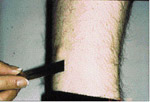
L’examen extra-buccale rťvťla des ostťomes au niveau frontal et ŗ la base de la mandibule, et ce, bilatťralement (ill. 6 et 7). Plusieurs lťsions cutanťes kystiques furent notťes, dont une ŗ la jambe droite (ill. 8). L’examen buccal dťmontra de nouveau la prťsence d’exostoses (ill. 9).
 |
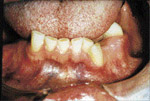 |
| Illustration 8 : Prťsence d’une lťsion kystique au niveau de la jambe droite du patient. Les kystes ťpidermoÔdes sont rarement observťs ŗ ces endroits, une caractťristique du syndrome de Gardner. | Illustration 9 : ņ l’examen buccal, on observe une exostose dans la rťgion prťmolaire gauche. |
Le patient ne rapportait aucun problŤme gastro-entťrologique en tant que tel, et aucun membre de sa famille n’en souffrait. Le patient fut rťfťrť en gastro-entťrologie, et la prťsence d’une polypose intestinale fut confirmť ŗ l’examen endoscopique. Par la suite, le patient subit une colectomie et est suivi rťguliŤrement pour la dťtection de lťsion maligne.
Conclusion
Nous avons prťsentť les caractťristiques particuliŤres des patients
atteints du syndrome de Gardner. La dťtection prťcoce de ce syndrome nous permettra de
diriger les patients vers des soins spťcialisťs. La prťsentation du second cas clinique
illustre bien le rŰle du dentiste comme professionnel de la santť pouvant agir
activement dans le cas d’une condition mťdicale extra-buccale. Le traitement
prťcoce de la condition intestinale du patient a ťvitť le dťveloppement fort probable
d’une lťsion cancťreuse.
Remerciements : Nous aimerions remercier M. Andrť Bťrard et Mme Chantale Boivin pour avoir aidť ŗ l’ťlaboration de cet article.
Le Dr Karazivan est prosthodontiste en pratique privťe ŗ Montrťal.
Le Dr Manoukian est dentiste, co-directeur du Dťpartement de mťdecine dentaire, Centre hospitalier de St-Mary, Montrťal.
Le Dr Lalonde est spťcialiste en mťdecine buccale, professeur agrťgť ŗ la Facultť de mťdecine dentaire de l’Universitť de Montrťal et directeur du programme de rťsidence multidisciplinaire (volet facultaire).
Demandes de tirťs ŗ part : Dr BenoÓt Lalonde, Facultť de mťdecine dentaire, Universitť de Montrťal, C.P. 6128, succursale Centre-Ville, Montrťal QC H3C 3J7.
Rťfťrences
1. Gardner EJ, Stephens FE. Cancer of the lower digestive tract in one family
group. Am J Hum Genet 1950; 2:41-8.
2. Gardner EJ, Richard RC. Multiple cutaneous and subcutaneous lesions occurring simultaneously with hereditary polyposis and osteomatosis. Am J Hum Genet 1953; 5:139-47.
3. Gorlin RJ, Cohen MM, Levin LS. Syndromes of the head and neck. 3rd ed. New York: Oxford University Press; 1990. p. 366-72.
4. Lothe RA, BrÝgger A, Gedde-Dahl T. High-resolution karyotypes of eighteen Norwegian polyposis patient. Cancer Genet Cytogenet 1990; 47:69-72.
5. Dhaliwal MK, Hughes JI, Jackson GL, Pathak S. Multiple polyposis coli associated with Gardner’s syndrome and chromosomal mosaicism: a family analysis. Am J Gastroenterol 1990; 85:880-3.
6. Rivera H, Simi P, Rossi S, Pardelli L, Di Paolo MC. A constitutional 5q23 deletion. J Med Genet 1990; 27:267-8.
7. Williams SC, Peller PJ. Gardner’s Syndrome. Case report and discussion of the manifestations of the disorder. Clin Nucl Med 1994; 19:668-70.
8. Kobayashi T, Narahara K, Yokoyama Y, Ueyama S, Mohri O, Fujii T and others. Gardner syndrome in a boy with interstitial deletion of the long arm of chromosome 5. Am J Med Genet 1991; 41:460-3.
9. Habif TP. Clinical dermatology — A color guide to diagnosis and therapy. 2nd ed. St-Louis: Mosby; 1990. p. 657.
10. Antoniades K, Eleftheriades I, Karakasis D. The Gardner syndrome. Int J Oral Maxillofac Surg 1987; 16:480-3.
11. Nakamura Y, Lathrop M, Leppert M, Dobbs M, Wasmuth J, Wolff E, and others. Localisation of the genetic defect in familial adenomatous polyposis within a small region of chromosome 5. Am J Hum Genet 1988; 43:638-44.
12. Jagelman DG, DeCosse JJ, Bussey HJ. Upper gastrointestinal cancer in familial adenomatous polyposis. Lancet 1988; 1(8595):1149-51.
13. Harrison’s principles of internal medicine. 14th ed. New York: McGraw-Hill Book Compagny; 1998. p. 572-3.
14. Dangel A, Meloni AM, Lynch HT, Sandberg AA. Deletion (5q) in a desmoid tumor of a patient with Gardner’s syndrome. Cancer Genet Cytogenet 1994; 78:94-8.
15. Itoh H, Ikeda S, Oohata Y, Iida M, Inoue T, Onitsuka H. Treatment of desmoid tumors in Gardner’s syndrome. Report of a case. Dis Colon Rectum 1988; 31:459-61.
16. Piffer S. Gardner’s syndrome and thyroid cancer — a case report and review of the litterature. Acta Oncol 1988; 27:413-5.
17. Wang M, Dobeck JM, Sorkin BC, Skobe Z. Adenomatous polyposis coli protein is expressed in alternate stages of the ameloblast life cycle. J Dent Res 1998; 77:1979-82.
18. Davies DR, Armstrong JG, Thakker N, Horner K, Guy SP, Clancy T, and others. Severe Gardner syndrome in families with mutations restricted to a specific region of the APC gene. Am J Hum Genet 1995; 57:1151-8.